



«Регуляторные исследования и экспертиза лекарственных средств» – научно-практический рецензируемый журнал открытого доступа, выпускаемый в печатной и онлайн-версиях. Основан в 1999 году.
Цель журнала: содействие развитию фармацевтической и медицинской науки и практики посредством опубликования и распространения информации о передовых достижениях в регуляторной деятельности и сфере обращения лекарственных средств.
Целевая аудитория. Предназначен для широкого круга специалистов, как российских, так и зарубежных, работающих в сфере обращения лекарственных средств:
- разработчиков и производителей лекарственных препаратов;
- представителей экспертных организаций, государственных регуляторных органов;
- работников контрольно-разрешительной системы и государственного надзора в сфере обращения лекарственных средств;
- сотрудников научно-исследовательских институтов, преподавателей, аспирантов и студентов медицинских, фармацевтических вузов, врачей и провизоров.
Более подробная информация – в разделе Цели и задачи.
Учредитель: ФГБУ «Научный центр экспертизы средств медицинского применения» Министерства здравоохранения Российской Федерации.
Периодичность: 6 раз в год.
Импакт-фактор: двухлетний импакт-фактор РИНЦ (2025) – 1,139.
Редколлегия. Географическое представительство:
- 8 стран
- 12 городов
Рецензирование:
- Двойное слепое
- Минимум 2 рецензента на рукопись
Основные метрики журнала:
14 дней в среднем от подачи до первого решения
88 дней в среднем от подачи до публикации в Интернете
15% приглашенных авторов
68% доля принятия рукописей
64 тыс. загрузок PDF в 2025 г.
Плата за публикацию: бесплатно.
Индексация. Входит в «Белый список» научных изданий (уровень 2), Перечень ВАК (категория К1), Russian Science Citation Index (RSCI), DOAJ. Информация об индексации в других российских и международных базах доступна в разделе «Индексирование».
Регистрация. Свидетельство о регистрации средства массовой информации ПИ № ФС77-82931 от 14 марта 2022 г.
Подписка. Подписной индекс в каталоге Пресса России – 57942, Урал-Пресс – 57942.
Текущий выпуск
ГЛАВНАЯ ТЕМА: ИННОВАЦИИ В ФАРМАЦЕВТИКЕ: ПУТЬ ОТ НАУЧНОЙ ИДЕИ К ТЕХНОЛОГИЧЕСКОМУ ЛИДЕРСТВУ

В интервью первый заместитель директора Центра трансфера медицинских технологий ФГБУ «НЦЭСМП» Минздрава России Д.И. Фёдорова рассказывает о практических инструментах, которые помогают управлять научно-технологическим развитием фармацевтической отрасли на основе данных. Центральной темой беседы является технологическое картирование, позволяющее системно анализировать направления исследований, уровень зрелости технологий и потенциал их внедрения. Поясняется, как этот инструмент может использоваться органами государственной власти и другими лицами, принимающими решения, для корректировки приоритетов исследований, распределения ресурсов и снижения рисков, а также какую ценность он представляет для фармацевтических и биотехнологических компаний при формировании портфеля разработок.

ВВЕДЕНИЕ. Стремительное развитие технологий генной терапии и расширение спектра их клинического применения опережают темпы адаптации нормативно-правовой базы. В Российской Федерации регулирование обращения препаратов клеточной и генной терапии характеризуется сложностью и фрагментарностью, обусловленной действием как национального законодательства, так и наднациональных норм Евразийского экономического союза.
ЦЕЛЬ. Анализ действующей нормативно-правовой базы, регулирующей жизненный цикл препаратов генной терапии в Российской Федерации (регистрация, производство, финансирование), выявление существующих правовых пробелов и коллизий, разработка рекомендаций по совершенствованию законодательства.
МАТЕРИАЛЫ И МЕТОДЫ. В работе с нормативно-правовыми актами (НПА) применяли системный и проблемный методологические подходы. Исследование состояло из четырех этапов, включавших контент-анализ нормативной базы с выделением ключевых понятий, сопоставление требований различных НПА для выявления зон неопределенности и коллизий, группировку проблем методами структурно-функционального и логического анализа и разработку рекомендаций на основе обобщенных данных.
РЕЗУЛЬТАТЫ. В работе проанализированы положения Федеральных законов № 61-ФЗ и 180-ФЗ, решения Совета Евразийской экономической комиссии, а также подзаконные акты, регулирующие правила GMP и программу государственных гарантий (ПГГ). Препараты генной терапии могут классифицироваться как высокотехнологичные лекарственные препараты, биотехнологические лекарственные препараты или биомедицинские клеточные продукты, что сопряжено с особенностями регулирования регистрации, производства и обеспечения пациентов лечением при наличии у препаратов соответствующих классификационных признаков. В статье обсуждаются ключевые различия в регулировании персонализированных методов терапии и продуктов, изготавливаемых на стандартизированной основе. В отношении персонализированных препаратов отсутствует законодательно определенная возможность централизованного производства на промышленных площадках с транспортировкой биоматериала, что ограничивает масштабирование технологий. Отмечена необходимость дополнительной работы по совершенствованию механизмов оплаты медицинской помощи с использованием методов генной терапии, не требующих регистрации, для расширения доступа пациентам к другим видам лечения наряду с уже включенными в ПГГ в рамках высокотехнологической медицинской помощи IV перечня.
ВЫВОДЫ. Для обеспечения доступности инновационной терапии целесообразны гармонизация законов № 61-ФЗ, № 180-ФЗ и Решения Совета ЕЭК № 78, унификация классификационных критериев препаратов генной терапии и создание нормативной базы для централизованного производства персонализированных продуктов, а также внедрение новых гибких механизмов тарификации для финансирования обеспечения пациентов.
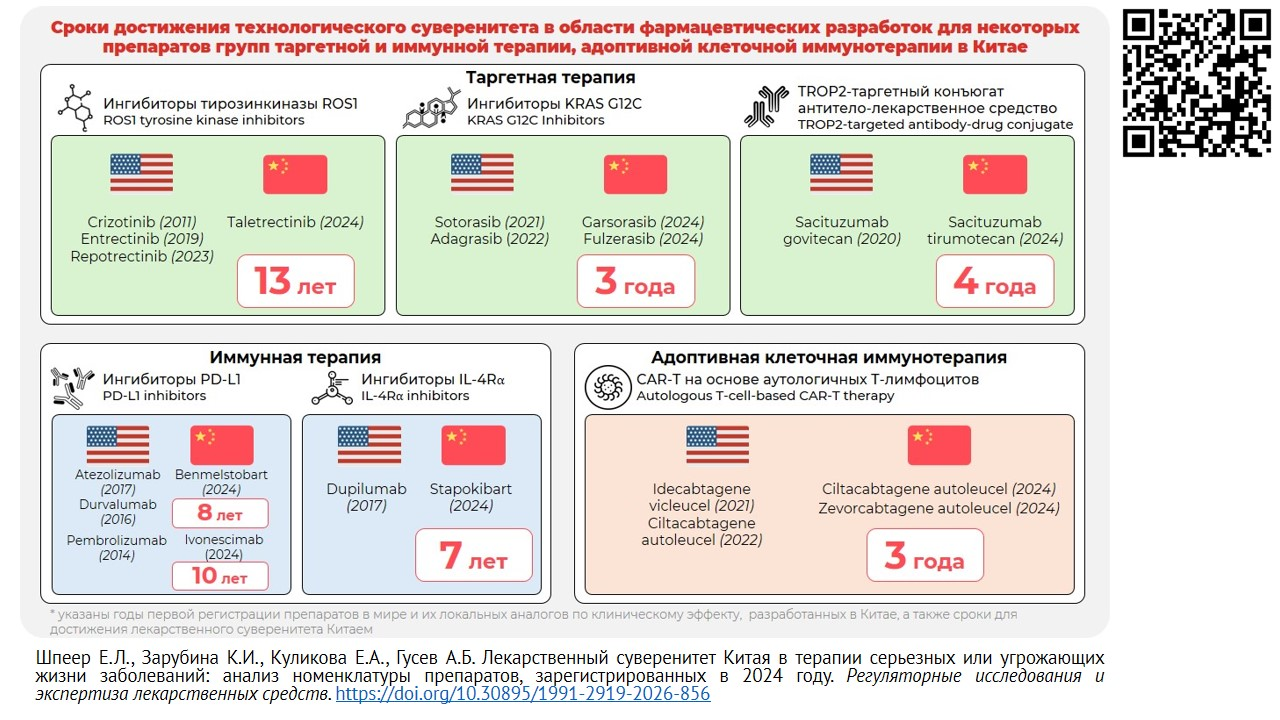
ВВЕДЕНИЕ. Деятельность дружественных государств по достижению и укреплению государственного суверенитета в области лекарственной безопасности востребована при формировании стратегии развития фармацевтической отрасли в Российской Федерации. В условиях ускоренного развития биомедицинских технологий и усиления роли национальных регуляторных механизмов представляет интерес опыт Китая по разработке и выводу инновационных лекарственных препаратов на локальный рынок.
ЦЕЛЬ. Анализ номенклатуры лекарственных препаратов 1-го класса по классификации Национальной администрации по медицинским продуктам КНР (NMPA), разработанных китайскими компаниями для терапии серьезных или угрожающих жизни заболеваний и зарегистрированных в Китае в 2024 г., для оценки их потенциала отнесения к first-in-class medicine, наличия ключевых технологических решений и потенциальной клинической значимости.
МАТЕРИАЛЫ И МЕТОДЫ. Оценка лекарственных препаратов, разработанных в Китае по программе «прорывной терапии» и зарегистрированных в 2024 г., проведена по данным отчета Национальной администрации по контролю за лекарственными средствами Китая (National Medical Products Administration, NMPA) за 2024 г.
РЕЗУЛЬТАТЫ. Сформирован перечень из 9 препаратов, отнесенных при регистрации к 1 классу, предназначенных для терапии серьезных или угрожающих жизни заболеваний, разработанных национальными компаниями, включая препараты таргетной терапии, иммунной терапии, конъюгаты антитело — лекарственное средство и CAR-T-препараты. Для каждого препарата проанализированы механизм действия, технологические особенности разработки, наличие зарубежных аналогов и клинические данные регистрационных исследований. Показано, что один препарат (ивонесцимаб) может быть отнесен к категории «первый в классе», тогда как остальные относятся к категории «следующий в классе». Для ряда препаратов (ивонесцимаб, талетректиниб, цилтакабтаген аутолейцел) выявлен потенциал достижения характеристик категории «лучший в классе» на основании данных пострегистрационного периода. Отмечено, что ключевые технологические решения направлены на повышение селективности, преодоление резистентности, улучшение фармакокинетических свойств и профиля безопасности.
ВЫВОДЫ. Результаты анализа свидетельствуют о формировании в Китае зрелой модели ускоренной разработки инновационных лекарственных препаратов с высоким потенциалом клинической значимости. Полученные данные могут представлять интерес для российских разработчиков в свете действующих национальных программ для достижения технологического суверенитета в области фармацевтических разработок.

ВВЕДЕНИЕ. Несмотря на широкое применение твердых лекарственных форм (ТЛФ) с модифицированным высвобождением (МВ), обобщающих работ, охватывающих полный цикл их разработки — от выбора технологической платформы до применения цифровых инструментов оптимизации, — в литературе выявлено немного. Имеющиеся публикации, как правило, рассматривают отдельные технологические платформы, не затрагивая вопросы цифровой оптимизации.
ЦЕЛЬ. Систематизация и критическая оценка современных подходов к оптимизации технологии получения твердых лекарственных форм с модифицированным высвобождением и определение роли цифровых инструментов в повышении эффективности фармацевтической разработки.
ОБСУЖДЕНИЕ. Анализ научных публикаций показал, что гидрофильные матрицы на основе гидроксипропилметилцеллюлозы (ГПМЦ) остаются ведущей платформой для разработки пролонгированных форм. Однако контроль высвобождения высокорастворимых субстанций требует комбинирования гидрофильных и гидрофобных полимеров. Осмотические системы обеспечивают pH-независимый профиль высвобождения, но их применение сопряжено с риском раздражения желудочно-кишечного тракта. Мультипартикулярные формы снижают вероятность непреднамеренного высвобождения всей дозы; при этом качество полимерного пленочного покрытия определяет воспроизводимость кинетики высвобождения. Экструзия горячего расплава в сочетании с 3D-печатью методом послойного наплавления (Fused Deposition Modeling, FDM) позволяет формировать заданный профиль высвобождения путем варьирования геометрии лекарственной формы. Установлено, что концепция встроенного качества (Quality by Design) и методы машинного обучения существенно сокращают объем экспериментальных работ, хотя проблемы интерпретируемости глубоких нейронных сетей и дефицит внешней валидации интеллектуальных моделей остаются нерешенными.
ЗАКЛЮЧЕНИЕ. Матричные системы на основе ГПМЦ занимают лидирующее положение среди технологий пролонгированного высвобождения благодаря масштабируемости и предсказуемой кинетике, однако оптимальный выбор технологии получения твердых лекарственных форм с модифицированным высвобождением определяется физико-химическими свойствами субстанции и фармакокинетическим профилем. Применение методов планирования эксперимента при разработке ТЛФ с МВ и машинного обучения позволяет сократить число экспериментальных работ. Перспективное направление дальнейших исследований связано с разработкой интерпретируемых прогнозных моделей и адаптацией нормативной базы к лекарственным формам, получаемым аддитивными методами (трехмерной печатью).
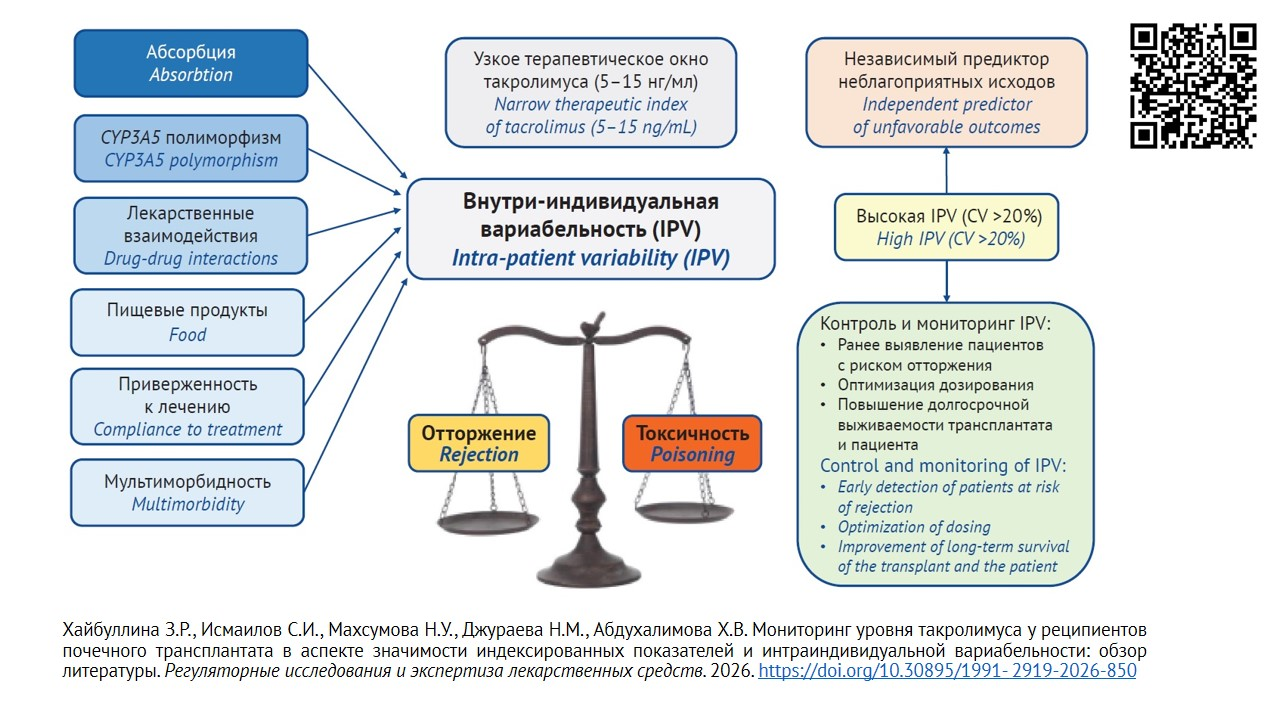
ВВЕДЕНИЕ. Такролимус (ТАС) в комбинации с микофенолата мофетилом и глюкокортикоидами составляют основу иммуносупрессивной терапии после трансплантации почки. Высокая интраиндивидуальная вариабельность концентрации такролимуса является индикатором нестабильности иммуносупрессии, что связано с риском отторжения трансплантата и требует постоянного мониторинга.
ЦЕЛЬ. Оценка методов определения уровня такролимуса в крови у реципиентов почечного трансплантата и сравнение различных подходов к мониторингу его концентрации.
ОБСУЖДЕНИЕ. Метод жидкостной хроматографии с масс-спектрометрией (ЖХ-МС/МС) является стандартом количественной оценки ТАС в различных видах биоматериала. В клинической практике внедрены иммунохимические методы его определения в цельной крови, однако они могут давать перекрестную реакцию с неактивными метаболитами ТАС, завышая получаемые результаты. Метод иммунохемилюминесценции на магнитных частицах обеспечивает надежность результата и позволяет определять концентрации ТАС до 0,5 нг/мл, а коэффициент вариации результата не превышает 15% в сравнении с референсным методом ЖХ-МС/МС. ТАС характеризуется узким терапевтическим окном, а его содержание у реципиентов почки как в крови (цельная кровь, плазма), так и в мононуклеарах крови отличается высокой интраиндивидуальной вариабельностью (IPV). Это обусловливает необходимость разработки новых подходов к оценке целевых уровней ТАС, включая выбор биоматериала и метода определения ТАС у пациентов из группы риска дисфункции почечного трансплантата. Наибольшую клиническую значимость из показателей эффективности иммуносупрессии имеет равновесная концентрация ТАС в крови, соотношение С/D предложено использовать для прогноза токсичности. Уменьшение этого соотношения может быть предиктором неблагоприятного прогноза ввиду токсичности ТАС у реципиентов почки. Показатель IPV напрямую отражает стабильность экспозиции препарата у конкретного пациента, позволяет оценить риск отторжения и токсичности. Высокая IPV является независимым предиктором неблагоприятных исходов у реципиентов почечного трансплантата. Величина IPV концентрации ТАС зависит от эндогенных и экзогенных факторов, таких как полиморфизм CYP3A5, особенности диеты, лекарственные взаимодействия, клинические ситуации. Регулярный контроль IPV и устранение влияющих факторов позволяют обеспечить хороший результат выживаемости трансплантата, как непосредственный, так и отдаленный.
ЗАКЛЮЧЕНИЕ. Такролимус характеризуется узким терапевтическим окном, а его содержание в крови у реципиентов почки отличается высокой интраиндивидуальной вариабельностью. Проведение межлабораторных сличений и разработка нормализованных показателей позволит минимизировать разночтения в оценке концентрации ТАС, а учет IPV позволит снизить риск неблагоприятных событий при краткосрочном и долгосрочном наблюдении реципиентов почки. Это обосновывает пересмотр подходов к мониторингу концентрации ТАС для улучшения результатов лечения, повышения выживаемости почечного трансплантата и реципиента.
ВВЕДЕНИЕ. Для реализации государственной стратегии «Фарма 2030» необходимо создание высокотехнологичных отечественных препаратов, в том числе бесклеточных тканеинженерных композиций, характеризующихся высокой эффективностью и низкой иммуногенностью. В рамках этой задачи проводится разработка и изучение фармакологических свойств спрея регенеративного действия для лечения ожоговых ран IIb–IIIa степени, который представляет собой тканеинженерную композицию из бесклеточного биоматериала пуповины человека и антимикробного компонента.
ЦЕЛЬ. Оценка регенеративной активности экспериментальных составов спрея, полученных на основе вартонова студня пуповины человека, на модели ожога у крыс.
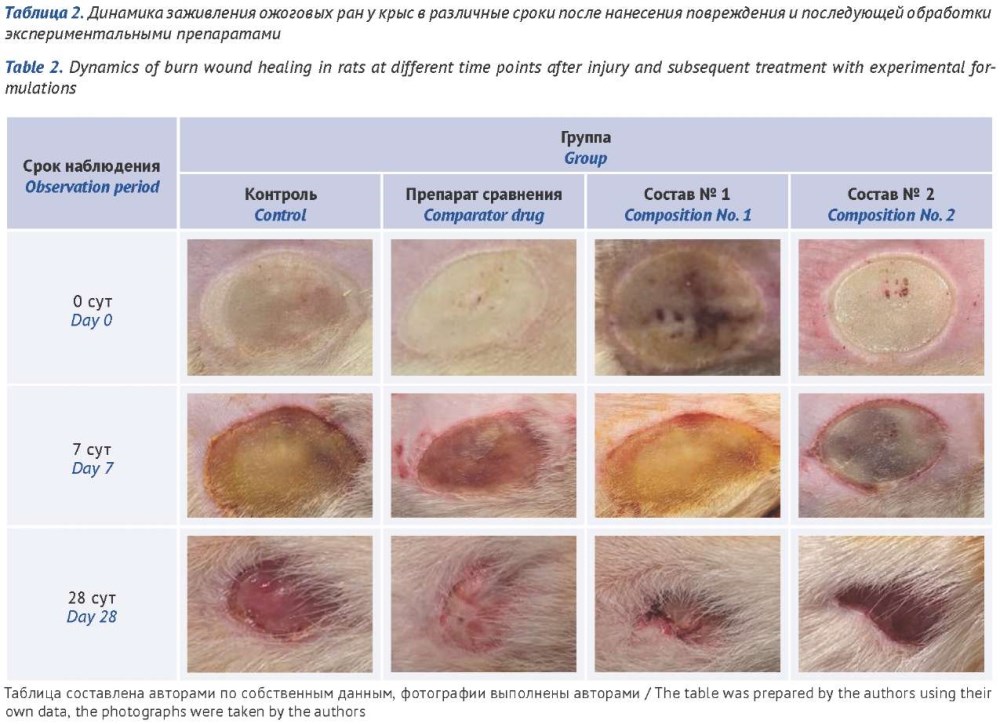
МАТЕРИАЛЫ И МЕТОДЫ. Разработанные составы спрея включали биодеградируемый лиофилизированный гидролизат бесклеточного матрикса вартонова студня пуповины человека и антибактериальные препараты: состав № 1 — с добавлением гентамицина сульфата, состав № 2 — неомицина сульфата. В качестве препарата сравнения использовали аэрозоль «Олазоль» («Алтайвитамины», Россия). Исследование проводили на модели глубокого ожога у взрослых крыс. Животные были разделены на группы, получавшие лечение экспериментальными составами спреев, препаратом сравнения, группу контроля (без лечения), группу интактных животных. Оценка регенеративной активности включала измерение площади раневого дефекта, а также концентрации лейкоцитов, С-реактивного белка (СРБ) и эпидермального фактора роста (ЭФР) при исследовании крови.
РЕЗУЛЬТАТЫ. К 28-м суткам наблюдения оба состава спрея статистически значимо превосходили препарат сравнения по уменьшению площади ожоговой раны: состав № 1 — на 66%, состав № 2 — на 20%. Применение экспериментальных составов спрея и препарата сравнения сопровождалось снижением уровня лейкоцитов и СРБ до значений, сопоставимых с таковыми у интактных животных. Концентрация ЭФР в экспериментальных группах статистически значимо повысилась: при лечении составом № 1 — на 104,0%, составом № 2 — на 76,5% относительно группы контроля и на 106,5 и 78,8% соответственно относительно группы интактных животных.
ВЫВОДЫ. Экспериментальные составы спрея, полученные на основе вартонова студня пуповины человека, обладают высокой регенеративной активностью, статистически значимо ускоряя заживление глубоких ожоговых ран и уменьшая выраженность воспалительного ответа у крыс. Результаты работы свидетельствуют о целесообразности их дальнейшего доклинического и клинического изучения с перспективой внедрения в клиническую комбустиологию.
КОНТРОЛЬ КАЧЕСТВА ЛЕКАРСТВЕННЫХ СРЕДСТВ
ВВЕДЕНИЕ. В Российской Федерации отсутствует фармакопейный стандарт на мед как фармацевтическую субстанцию, а действующие нормативные документы (ГОСТ 19792-2017, зарубежные фармакопеи) предъявляют различные требования к его качеству, которые отличаются по контролируемым показателям, методикам и нормам. Для разработки единых подходов к фармакопейной стандартизации актуальным представляется проведение сравнительного анализа национальных и зарубежных требований к качеству меда.
ЦЕЛЬ. Сравнительный анализ показателей качества меда, регламентируемых зарубежными фармакопеями и ГОСТ 19792-2017, для обоснования перечня критических показателей, который может быть использован при разработке фармакопейной статьи на мед как фармацевтическую субстанцию.
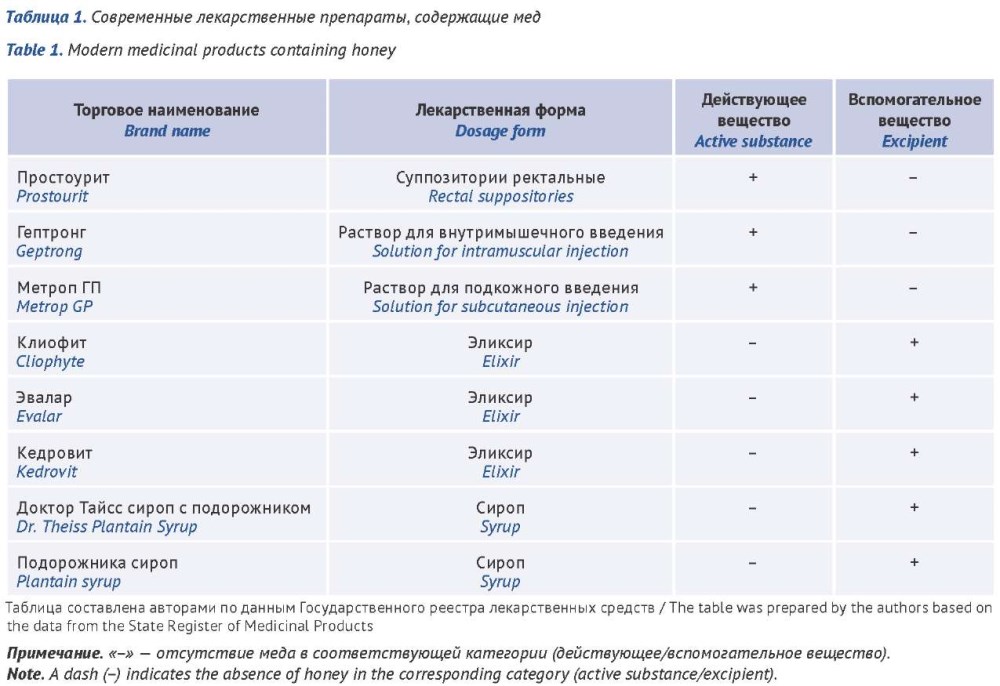
ОБСУЖДЕНИЕ. Проведен обзор источников литературы в базах данных PubMed, Google Scholar, eLIBRARY.RU за 2015–2025 гг. Проведен сравнительный анализ действующих монографий на мед в Европейской (Ph. Eur.), Корейской (КP), Китайской (ChP), Японской (JP) фармакопеях и Фармакопее США (USP), а также ГОСТ 19792-2017 (ГОСТ). В Российской Федерации зарегистрировано 8 лекарственных препаратов (ЛП), содержащих мед, причем в трех из них он выступает как действующее вещество, в пяти — как вспомогательное. Сравнительный анализ монографий выявил существенные различия в подходах к стандартизации меда как фармацевтической субстанции. В Ph. Eur. подлинность подтверждается по профилю сахаров методом тонкослойной хроматографии (ТСХ), в USP — качественной реакцией на пролин. Содержание глюкозы, фруктозы и их соотношение определяют только в ChP и ГОСТ. Наиболее критические расхождения обнаружены для показателей термической обработки меда и его фальсификации: нормы для 5‑гидроксиметилфурфурола (5-ГМФ) от 25 млн–1 в ГОСТ до 80 ppm в Ph. Eur. и KP, а перечень контролируемых примесей в ChP, JP и ГОСТ не совпадает. Контроль примесей, свидетельствующих о фальсификации, предусмотрен в ChP, JP и ГОСТ, но критерии отличаются: качественная реакция с йодом на крахмал и декстрин (ChP, KP, JP), реакция с таниновой кислотой (KP, JP), ТСХ на олигосахариды и ВЭЖХ на сахарозу/мальтозу (ChP), массовая доля сахарозы (ГОСТ). Определение диастазного числа предусмотрено только ГОСТ. Зарубежные производители ЛП ориентируются на требования Ph. Eur., отечественные — на ГОСТ и внутренние спецификации, что создает препятствия для гармонизации. Полученные данные обосновывают необходимость унификации требований к меду как фармацевтической субстанции и пересмотра действующих нормативных документов.
ВЫВОДЫ. Впервые проведен системный сравнительный анализ требований российских и зарубежных нормативных документов к качеству меда, что позволило выявить критические расхождения и установить наиболее значимый перечень показателей качества меда, требующих гармонизации, сформировать перспективный перечень показателей качества, который может быть использован при разработке фармакопейной статьи на мед как фармацевтическую субстанцию.
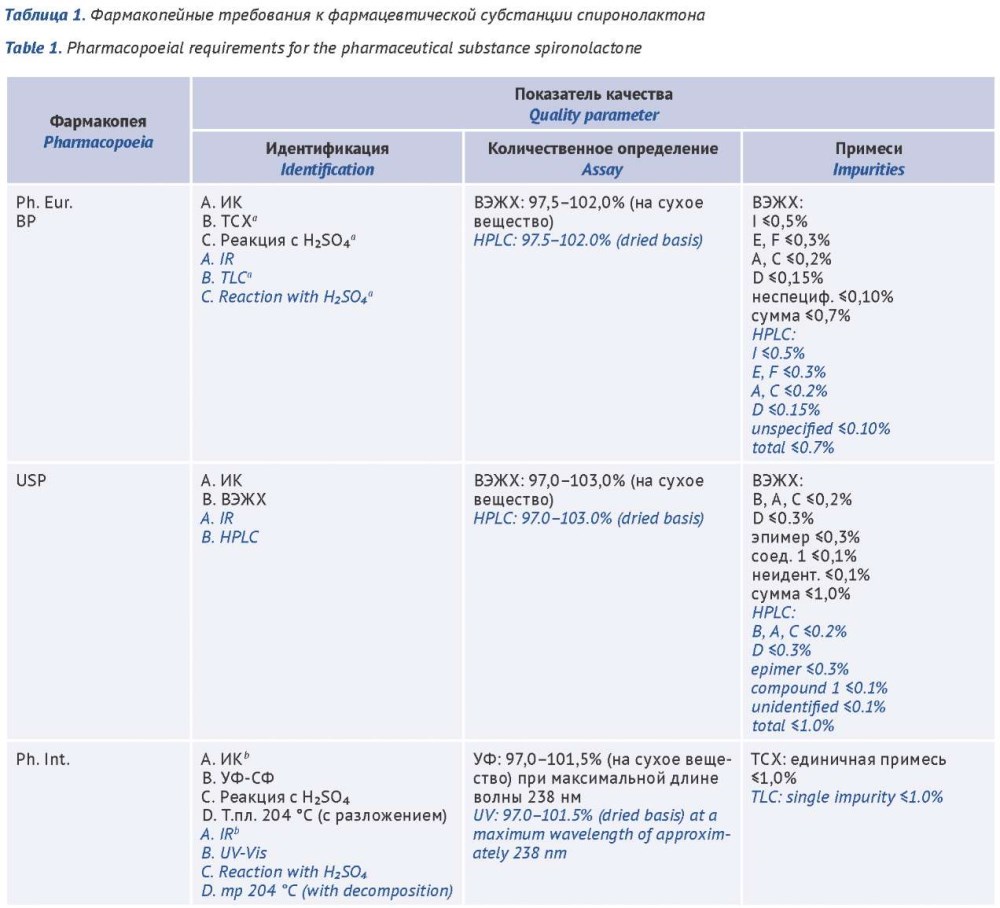
ВВЕДЕНИЕ. Спиронолактон, включенный в перечень жизненно необходимых и важнейших лекарственных препаратов, применяется при хронической сердечной недостаточности, артериальной гипертензии и первичном гиперальдостеронизме, что обусловливает высокие требования к качеству его субстанции и лекарственных форм. В действующих фармакопейных требованиях на спиронолактон имеются существенные различия в подходах к контролю примесей, идентификации и количественному определению, что создает регуляторные барьеры и осложняет оценку качества препаратов. Для решения этой проблемы авторами проведен сравнительный анализ требований фармакопей с целью выявления направлений гармонизации.
ЦЕЛЬ. Сравнительная оценка требований зарубежных фармакопей и Государственной фармакопеи Российской Федерации к качеству фармацевтической субстанции и таблетированных форм спиронолактона для определения направлений гармонизации национальных фармакопейных статей.
ОБСУЖДЕНИЕ. Объектами анализа послужили действующие монографии Европейской фармакопеи (Ph. Eur.), Фармакопеи США (USP), Британской фармакопеи (BP), Индийской фармакопеи (IP), Японской фармакопеи (JP), Китайской фармакопеи (ChP), Международной фармакопеи (Ph. Int.), Корейской фармакопеи (KP) и Государственной фармакопеи Российской Федерации (ГФ РФ). Методология основана на сравнительном анализе монографий по ключевым показателям качества: «Идентификация», «Количественное определение», «Родственные примеси», «Растворение» (для таблеток). Сравнительный анализ показал, что наиболее жесткие и детализированные требования к родственным примесям предъявляют Ph. Eur. и USP, где определение осуществляется методом ВЭЖХ. В ряде других фармакопей (ГФ РФ, IP, ChP) сохраняются менее селективные методы (УФ-спектрофотометрия, тонкослойная хроматография) и обобщенные критерии приемлемости для примесей, что не позволяет в полной мере контролировать стабильность и чистоту субстанции. Для лекарственных форм выявлена высокая степень гармонизации условий теста «Растворение» при сохраняющихся различиях в критериях приемлемости и методиках количественного анализа. Приоритетным направлением развития национальных фармакопейных статей являются внедрение хроматографических методов и гармонизация норм содержания примесей с ведущими международными стандартами.
ЗАКЛЮЧЕНИЕ. Выявленные различия в фармакопейных подходах подтверждают целесообразность пересмотра национальных стандартов в сторону их гармонизации с требованиями Ph. Eur. и USP. Включение высокоспецифичного метода ВЭЖХ и детализированного нормирования примесей в фармакопейные статьи ГФ РФ на лекарственные средства спиронолактона будет способствовать обеспечению качества и безопасности препаратов.
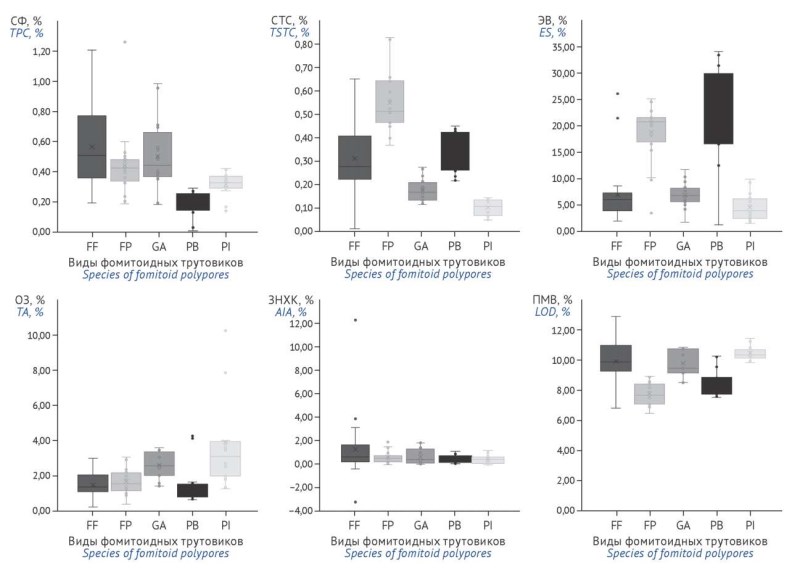
ВВЕДЕНИЕ. Актуальность настоящего исследования обусловлена возрастающим интересом к трутовым грибам как доступному и возобновляемому источнику биологически активных веществ с доказанной фармакологической активностью (прежде всего фенольных и тритерпеновых соединений). Несмотря на широкую распространенность фомитоидных трутовиков, их сырье практически не стандартизовано и не представлено в фармакопеях большинства стран, что ограничивает его внедрение в медицинскую практику. Отсутствие унифицированных подходов к оценке качества и выбору аналитических маркеров, с учетом высокой вариабельности химического состава, затрудняет стандартизацию сырья. В связи с этим обоснование критериев качества и методов стандартизации сырья фомитоидных трутовиков является важной задачей.
ЦЕЛЬ. Обоснование критериев качества сырья фомитоидных трутовиков для последующей разработки нормативного документа.
МАТЕРИАЛЫ И МЕТОДЫ. Исследовано не менее 20 партий плодовых тел фомитоидных трутовиков видов Fomes fomentarius (L.) Fr., Fomitopsis pinicola (Sw.) P. Karst., Ganoderma applanatum (Pers.) Pat., Piptoporus betulinus (Bull.) P. Karst., Phellinus igniarius (L.) Quél., заготовленных в различных регионах Республики Беларусь и Российской Федерации. Определены потеря в массе при высушивании, общая зола, не растворимая в хлороводородной кислоте зола, сумма экстрактивных веществ, сумма фенольных соединений и сумма стероидных и тритерпеновых соединений спектрофотометрическим и гравиметрическим методами.
РЕЗУЛЬТАТЫ. Показана межвидовая и внутривидовая вариабельность показателей качества. Установлено, что в зависимости от вида сырья допустимые пределы содержания суммы фенольных соединений составляют 0,10–0,25%, суммы стероидных и тритерпеновых соединений — 0,05–0,30%, экстрактивных веществ — 0,5–14,0%. Показатели общей золы, не растворимой в хлороводородной кислоте золы и потери в массе при высушивании находятся в пределах 1,0–5,0, 1,0–3,0 и 10,0–12,0% соответственно.
ВЫВОДЫ. Разработаны показатели качества сырья фомитоидных трутовиков, которые могут быть использованы для создания нормативного документа по качеству на сырье грибного происхождения.
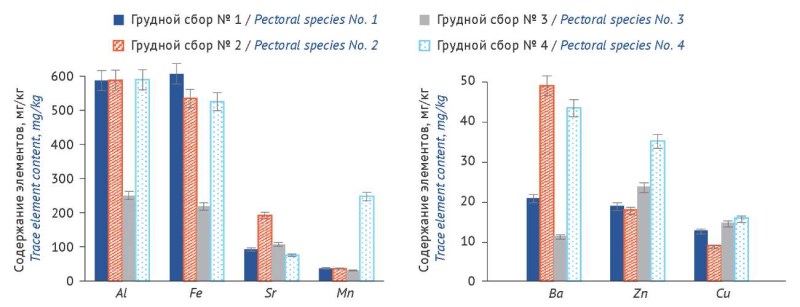
ВВЕДЕНИЕ. Микроэлементы способны влиять на фармакологический эффект лекарственных растительных препаратов, однако контроль их качества в настоящее время ограничивается оценкой содержания только токсичных элементов (Pb, Cd, Hg, As) без изучения полного микроэлементного профиля. Исследования элементного состава комплексных растительных препаратов, представленных на фармацевтическом рынке, является актуальным.
ЦЕЛЬ. Провести сравнительную оценку содержания микроэлементов (Al, Ba, Cu, Fe, Mn, Sr, Zn) в фармакопейных грудных сборах.
МАТЕРИАЛЫ И МЕТОДЫ. Объекты исследования — образцы грудных сборов № 1, 2, 3, 4 российских производителей. Методом атомно-эмиссионной спектрометрии с индуктивно связанной плазмой проведен анализ 7 элементов, указанных в цели работы.
РЕЗУЛЬТАТЫ. Содержание (мг/кг) микроэлементов в образцах грудных сборов варьировало в диапазонах: Fe 134,9–707,8; Mn 25,6–248,7; Cu 3,8–16,9; Zn 4,3–35,7; Al 184,2–769,8; Ba 8,4–43,8; Sr 74,3–254,6. Обнаружено, что элементы различались по уровню концентраций: Fe, Al — высокий; Sr, Mn — средний; Ba, Cu, Zn — низкий. Расчетное суточное поступление в организм человека составило: Ba 0,08–0,43 мг/сутки; Cu 0,03–0,16 мг/сутки, что в 3–18 и 19–100 раз соответственно ниже установленных допустимых суточных уровней (1,4 и 3,0 мг/сут), установленных Решением Коллегии Евразийской экономической комиссии № 138.
ВЫВОДЫ. Впервые проведен сравнительный анализ содержания микроэлементов (Al, Ba, Cu, Fe, Mn, Sr, Zn) в грудных сборах № 1–4 разных производителей. На фрагменте микроэлементного профиля показано, что содержание микроэлементов в отдельных сборах различалось в 3–8 раз в зависимости от источников сырья, выбранных производителем. Выявлены характерные особенности максимального накопления элементов в каждом из них: грудной сбор № 1 — Fe; грудной сбор № 2 — Sr; грудной сбор № 4 — Mn, Cu и Zn. Это следует учитывать при использовании фитотерапии для лечения и профилактики заболеваний верхних дыхательных путей.
Объявления
2026-07-07
Внимание конкурс!
Редакция журнала «Регуляторные исследования и экспертиза лекарственных средств» информирует о проведении ежегодного конкурса «Статья года».
| Еще объявления... |

ISSN 3034-3453 (Online)





























